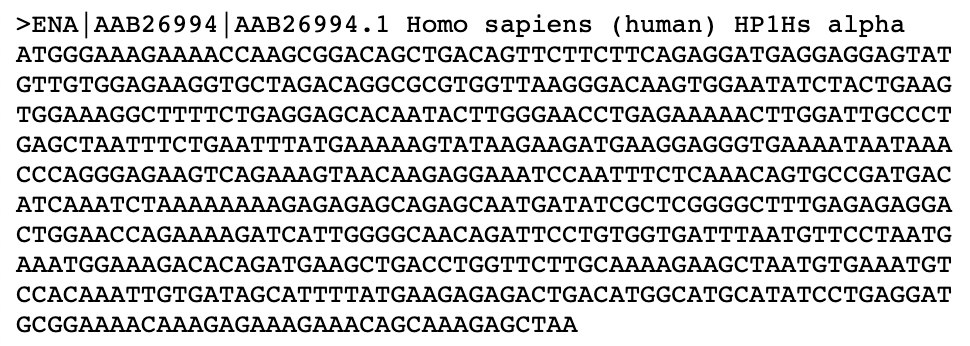
过夜生长在琼脂糖- LB平板上的菌落,过目可知。也许你会很惊讶,为什么LIC后的平板上只有可怜的几个菌落?是的,这是正常的。根据我们的经验,这些单一菌落是目的质粒的可能性非常大。相反,如果你的琼脂糖- LB平板上到处是菌落,那这些菌落是假阳性的概率很高。能够生长在这些含有抗生素的平板上的菌落必定含有抗性基因,因此,这些假阳性菌落多半是含有template的质粒。进一步分析,应该是Bpn1消化过程中出现了差错。菌落PCR是检验这些菌落是否含有insert的直接方法。菌落PCR的DNA模板就是这些菌落——将移液器的枪头在标记好的菌落上点一下(挑取),然后吹打进PCR反应体系中即可。标记菌落的目的是为了在下次寻找相应的阳性菌落的时候,简单快捷。菌落PCR的引物就是insert PCR的引物,无需额外定制引物。菌落PCR的反应条件(退火温度,延伸时间)与insert PCR的一致。细心的小伙伴甚至会用提供template的质粒作为隐性对照,以提供insert的质粒作为阳性对照。总结的时候,将template PCR, insert PCR 和菌落PCR并排放在一起的时候,完成的LIC实验过程就出来了,如图一。
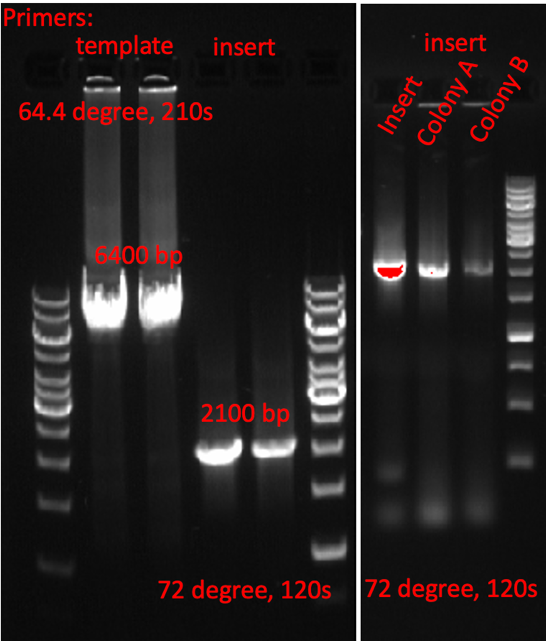
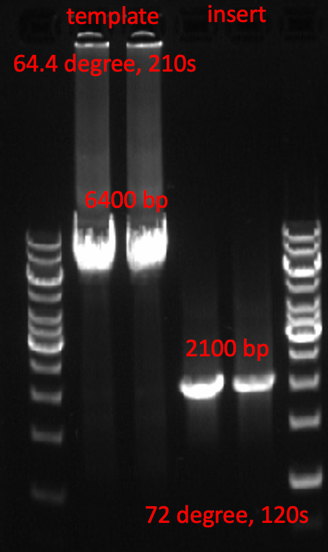
图一. Template PCR, insert PCR 和菌落PCR的电泳结果。选取的两个菌落都是阳性。
当平板上有大量菌落时,应该多挑取一些菌落作PCR。如下图,我们挑取了7个菌落做PCR,但是,只有#4 菌落含有insert。(这里能检测出#4,含有运气成分。)
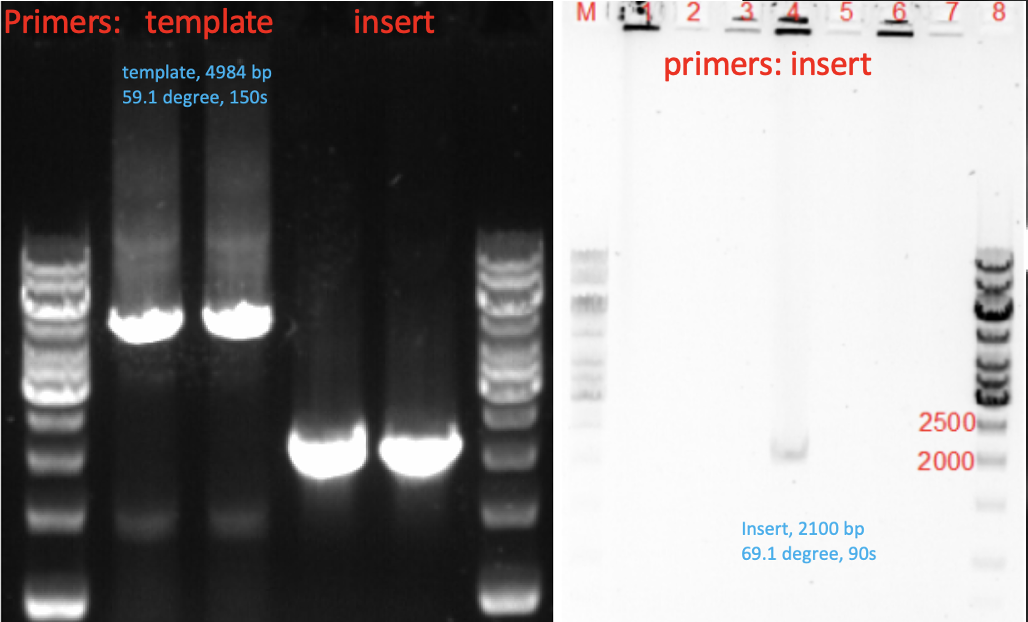
图二. Template PCR, insert PCR和菌落PCR的电泳结果。选取的7个菌落,只有一个是阳性。
LIC的反应体系是10 ul,而一个转化反应通常只需要5 ul。因此,为了避免浪费,5 ul可以转化到DH5a 中,专门用来提取目的质粒;另外5 ul转化到BL21 (升级版本的叫作Rosetta)中,专门用来做目的蛋白的表达和纯化。